


Susanna M. Hamilton/Broad Institute
Science & Tech
New details about mumps outbreaks of 2016‒17
Studying mumps virus genomes filled in gaps about its spread across campuses
In 2016 and 2017, a surge of mumps cases at Boston-area universities prompted researchers at the Broad Institute of MIT and Harvard to study mumps virus transmission using genomic data, in collaboration with the Massachusetts Department of Public Health and local university health services.
As the outbreaks unfolded, the teams analyzed mumps virus genomes collected from patients, revealing new links between cases that first appeared unrelated and other details about how the disease was spreading that weren’t apparent from the epidemiological investigation.
The teams shared their sequencing data and findings in real time during the outbreaks, with both each other and the broader scientific community, and now report their conclusions in PLOS Biology.
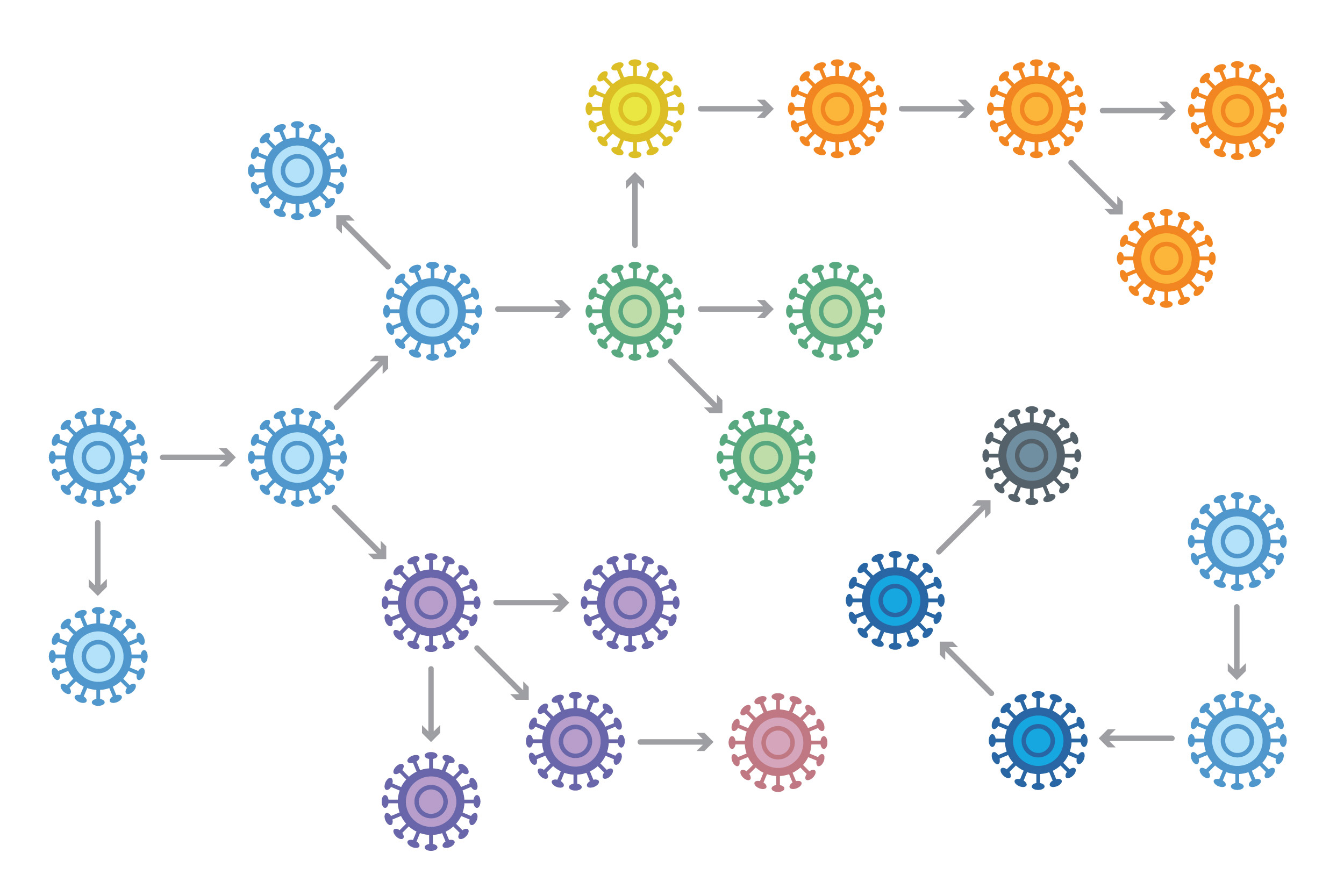
Analyzing viral genomes from an outbreak can show how a virus is evolving and being transmitted — data that can help public-health officials slow and stop the spread of disease.
“High-resolution genomic data about a virus, gathered from patient samples, allows us to reconstruct parts of an outbreak that aren’t evident at first,” said co-senior author Pardis Sabeti, Broad Institute member, professor at Harvard University, and Howard Hughes Medical Institute investigator. “The better we understand transmission chains in situations like this, the better we can inform efforts to control outbreaks and devise strategies to predict and stop them in the future.”
In Massachusetts, the typical rate of mumps is less than 10 cases per year — but more than 250 cases were reported in 2016 and more than 170 in 2017, despite high rates of vaccination. Many of the cases were from 18 colleges and universities in the state, including Harvard University, University of Massachusetts Amherst, and Boston University (these three universities met certain criteria to ensure patient privacy protection in this study and agreed to be named in the paper). Other outbreaks flared elsewhere in Boston and across the country around the same time.
The researchers found a clear link between cases at Harvard and an outbreak in East Boston, which were classified as distinct outbreaks during the initial public health investigation.
These patterns of cases raised questions about how much the virus was circulating in the Massachusetts and U.S. populations. To learn more, the research teams paired traditional epidemiological data with analysis of mumps virus whole-genome sequences from 201 infected individuals, focusing primarily on the Massachusetts university communities.
Mumps insight
The viral genomic data revealed details about the Boston-area outbreaks that could not be reconstructed by relying solely on more traditional approaches. For example, the researchers found a clear link between cases at Harvard and an outbreak in East Boston, which were classified as distinct outbreaks during the initial public health investigation.
Public health officials first thought the cases in these two communities were unrelated based on several pieces of evidence: epidemiological data, the different demographic makeup of the two populations (older adults with no obvious University connection versus mostly college-aged students), and a long gap between the apparent end of the outbreak at Harvard and the cases in the local community.
However, the genomic data indicated that the mumps viruses in the East Boston cases were genetically similar to those in the Harvard virus samples. This finding enabled the teams to identify contacts and transmission links between the University and the wider community.
“Even though the two outbreaks were occurring at different places and different times, we were able to show connections between these outbreaks that were operationally informative,” explained senior co-author Bronwyn MacInnis, associate director of malaria and viral genomics in the Infectious Disease and Microbiome Program and co-lead of the Global Health Initiative at Broad. “The public health teams could determine that they were essentially dealing with one problem, not two.”
In Massachusetts, the typical rate of mumps is less than 10 cases per year — but more than 250 cases were reported in 2016 and more than 170 in 2017, despite high rates of vaccination.
Understanding such transmission routes can help guide the outbreak response — for example, by determining whether efforts should be focused more on controlling transmission within a single community or between different ones.
“Whole-genome sequencing of patient samples helps us reconstruct the progression of an outbreak,” said co-first author Shirlee Wohl, formerly a Harvard graduate student in the Sabeti lab and now a postdoctoral fellow at Johns Hopkins University. “Traditional outbreak surveillance efforts can help identify possible sources of infection, but whole-genome sequencing can confirm these links and even suggest new, unexplored connections.”
The team emphasized that this study was made possible by the close partnerships it had with the Massachusetts Department of Health and the health-services teams at several universities. “I am proud to be part of the Massachusetts higher-education community,” Sabeti added. “They worked together and demonstrated the necessity of transparency in outbreak response. This is not a story of mumps at these universities, but of outstanding mumps reporting.”
Mutating mumps?
Another question of particular interest to the local teams was whether a new mutation in the mumps virus — for example, one that allows it to evade the immune system in a vaccinated individual — might have sparked the outbreak. Of the infected individuals, 65 percent had received the recommended two doses of the MMR vaccine. However, given the available data, the researchers found no evidence that genetic variants arising specifically during this outbreak contributed to the disease spread. This finding suggests that, in the Boston area, the virus wasn’t evolving into one that could dodge vaccine-induced immunity.

In addition to the findings related to the Boston-area outbreaks, the study’s broader geographic analysis suggested that the mumps virus has been circulating continuously at a low rate around the U.S., only rarely flaring up into notable outbreaks as in 2016 and 2017.
“This whole endeavor demonstrated the value of genetic data to the epidemiological health response, and of data-sharing among collaborating teams,” Sabeti said. “One of our goals is to build this capacity in many areas around the world so that public-health officials can rapidly mobilize and do this type of analysis whenever they need to.”
Co-first authors on this study include Hayden Metsky (Broad Institute, MIT) and Stephen Schaffner (Broad Institute, Harvard University, Harvard T.H. Chan School of Public Health). Co-senior authors on this study are Paul Rota (CDC), Lawrence Madoff (Massachusetts DPH, University of Massachusetts Medical School), Nathan Yozwiak (formerly Broad Institute/Sabeti lab), Bronwyn MacInnis, Sandra Smole (Massachusetts DPH), Yonatan Grad (Broad Institute, Harvard T.H. Chan School of Public Health), and Pardis Sabeti.
Support for this study was provided by the National Institute of Allergy and Infectious Diseases (U19AI110818, U54GM088558), Howard Hughes Medical Institute, a Harvard University Burke Global Health Fellowship, Amazon Web Services Cloud Credits for Research, and the National Institute of General Medical Sciences (T32GM007753).





