


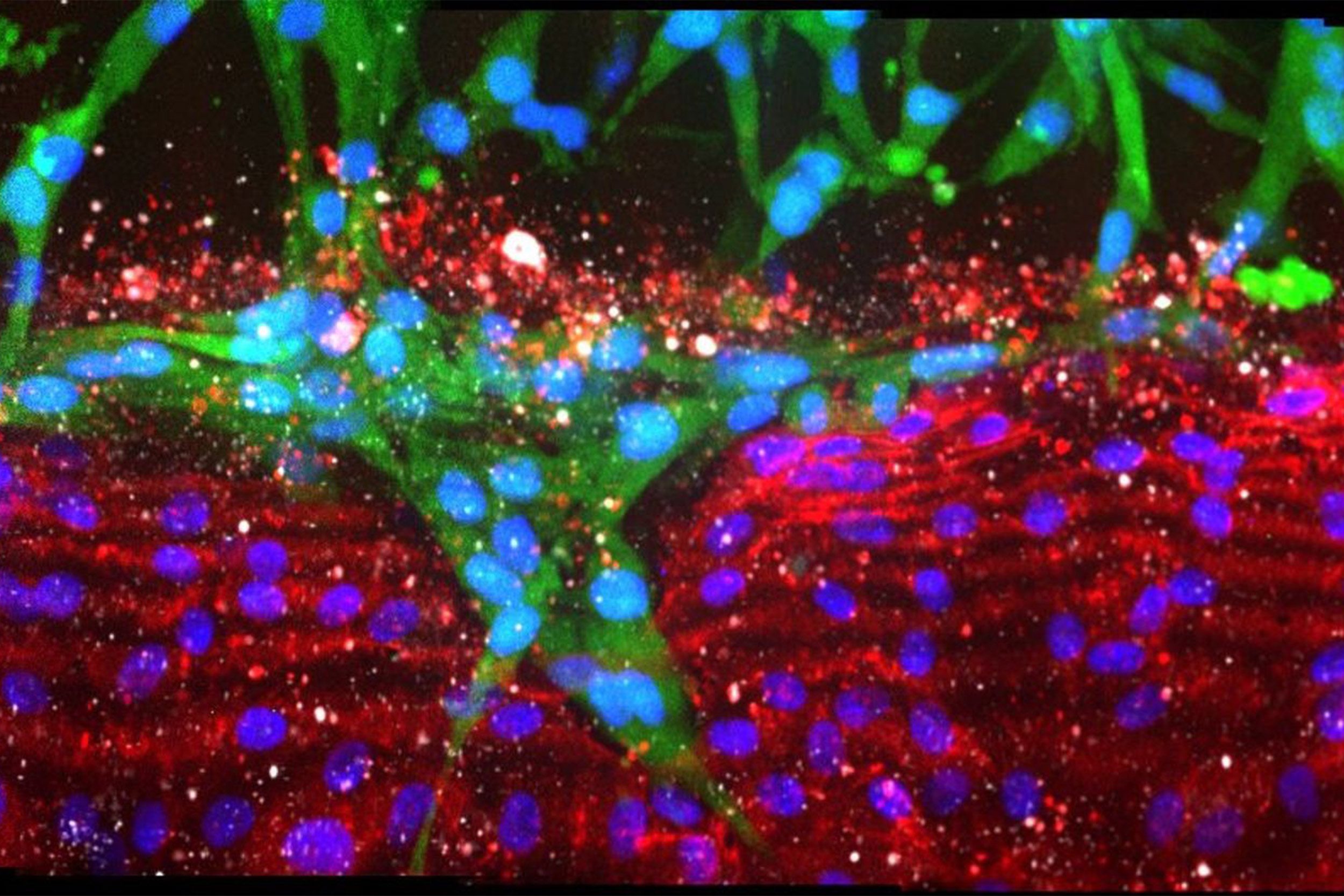
Pancreatic cancer cells (green) ablating endothelial cells lining a blood vessel channel (red), replacing healthy cells with tumor cells.
Science & Tech
Exposing how pancreatic cancer does its dirty work
Organ-on-a-chip study reveals mechanism that destroys and replaces nearby blood vessels, driving malignancy
Pancreatic cancer is one of the most insidious forms of the disease, with an average of only 9 percent of patients alive five years after diagnosis. One of the reasons for this dismal outcome is that pancreatic cancer cells are able to escape from tumors and enter the bloodstream very early in the disease course, meaning that by the time the cancer is discovered, usually it has already spread. Paradoxically, pancreatic tumors appear to almost lack blood vessels altogether, which prevents cancer drugs from reaching and killing them and has puzzled scientists and clinicians trying to understand how the disease progresses.
A new study from Harvard’s Wyss Institute for Biologically Inspired Engineering, Boston University, and the University of Pennsylvania has finally shed light on this mystery. Using both in vitro and in vivo models of pancreatic cancer and vasculature, it found that the tumor cells invade nearby blood vessels, destroy the endothelial cells that line them, and replace those cells with tumor-lined structures. This process seems to be driven by the interaction between the protein receptor ALK7 and the protein Activin in pancreatic cancer cells, pointing to a possible target for future treatments. The research is published in Science Advances.
Identifying the weapon
“Our study really brings to light the importance of ‘rescuing’ the vasculature prior to treating pancreatic cancer, because this disease is actively destroying our only route for delivering drugs to metastatic tumors,” said co-first author Duc-Huy Nguyen, a postdoctoral associate at Weill Cornell Medicine who performed the research while a graduate student at the University of Pennsylvania. “If we could prevent the cancer’s ablation of the surrounding endothelium by developing an inhibitor specific to the ALK7-Activin pathway, we could preserve the existing blood vessels and deliver drugs to patients to shrink down the tumor mass, which it is currently impossible to do.”
Catching a killer
Studying the interactions between pancreatic cancer and blood vessels historically has been very difficult, as it would require multiple, invasive tissue biopsies from human cancer patients, and imaging the disease over time in the internal organs of living mouse models is technically very challenging. The researchers took a different approach by using organs-on-chips: clear, flexible, plastic chips about the size of a USB stick containing microfluidic channels embedded in a collagen matrix that can be lined with living cells kept alive via a constant flow of nutrient-rich media.
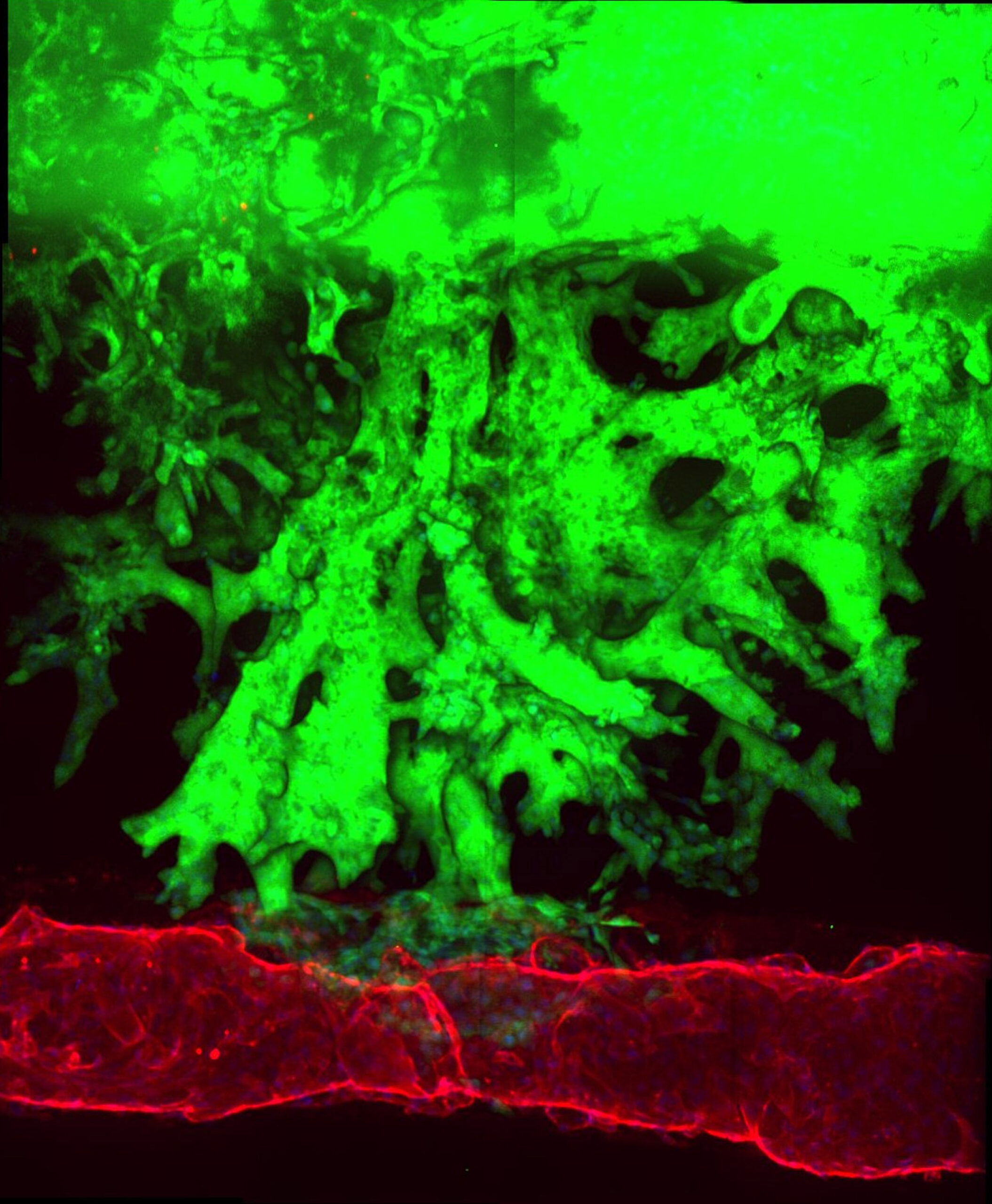
To replicate a pancreatic cancer tumor, the team seeded one channel with mouse pancreatic cancer cells, and a neighboring channel with human endothelial cells. They observed that after about four days, the pancreatic cancer cells began to invade the collagen matrix toward the blood vessel channel, eventually wrapped themselves around the channel, spread along its length, and finally invaded it. During the invasion process, the endothelial cells in direct contact with the cancer cells underwent apoptosis (cell death), leaving a blood vessel channel composed exclusively of cancer cells. They saw the same pattern when using human pancreatic cancer cells in the organ-on-a-chip, and in living mouse models of pancreatic cancer, suggesting that this process may also occur in humans.
The researchers suspected that the mechanism by which pancreatic cancer cells ablate endothelial cells had something to do with the TGF-β signaling pathway, a cascade of molecular interactions that has been implicated in multiple types of cancers. They introduced a TGF-β inhibitor into their organ-on-a-chip cancer model for seven days, and saw that the ablation of endothelial cells was significantly reduced. When pancreatic cancer cells were implanted into mice that were subsequently given the same inhibitor molecule, their tumors displayed a higher density of blood vessels, confirming that the inhibitor also reduced ablation in vivo.
To further hone in on the specific TGF-β receptor(s) driving the ablation process, the team created a co-culture device in which they grew pancreatic cells surrounded by endothelial cells so they could investigate exactly what was happening at the interface between the two cell types. They identified three candidate receptors — ALK4, ALK5, and ALK7 — and genetically deleted the gene coding for each receptor, first in the endothelial cells, then in the pancreatic cancer cells. They found that only by deleting ALK7 from the cancer cells could they significantly reduce ablation of endothelial cells and slow cancer cell growth.
The ALK7 receptor has two known binding partners, the proteins Activin and Nodal, and when the researchers exposed in vitro cancer cells to compounds that inhibit each partner, only the Activin inhibitor reduced endothelial ablation, suggesting that the interaction between ALK7 and Activin is the major driver of pancreatic cancer’s growth and metastasis. This was further confirmed by knocking out ALK7 expression in cancer cells and then implanting them into mice, which resulted in slower-growing in vivo tumors with higher blood-vessel density and fewer apoptotic endothelial cells.
“Not only has our study revealed a major insight into pancreatic cancer biology that could be used to drive the development of new treatments, our cancer-on-a-chip platform opens a new door to being able to more carefully study the interactions between blood vessels and other types of cancers, which could be extremely useful in teasing out these important but complex interactions,” said co-first author Esak (Isaac) Lee, who was a postdoctoral fellow at the Wyss Institute and Boston University when the research was carried out and is now an assistant professor at Cornell University.
The team is looking into developing their platform to further understand additional cellular interactions in cancer, including between cancer and immune cells, and between cancer and the perivascular cells that surround and support blood vessels.
“This study really demonstrates the power of using 3D and 2D organotypic models to replicate disease states in vitro and identify precise mechanisms, and their superiority over traditional in vitro and in vivo approaches,” said corresponding author Chris Chen, an associate faculty member at the Wyss Institute who is also a professor of biomedical engineering and director of the Tissue Microfabrication Laboratory at Boston University. “We are really just beginning to scratch the surface, and we’re excited to see what other kinds of insights we can uncover with this platform that could lead to new and better treatments.”
Additional authors of the paper include former Wyss Institute postdoctoral researcher Styliani Alimpert; Jeroen Eyckmans at the Wyss Institute and Boston University; Robert Norgard and Ben Stanger at the Perelman School of Medicine at the University of Pennsylvania; former Boston University graduate student Alec Wong; and Jake June-Koo Lee at Harvard Medical School.
This research was supported by the National Institutes of Health, the Lymphatic Education and Research Network, and the Ludwig Center at Harvard.





