


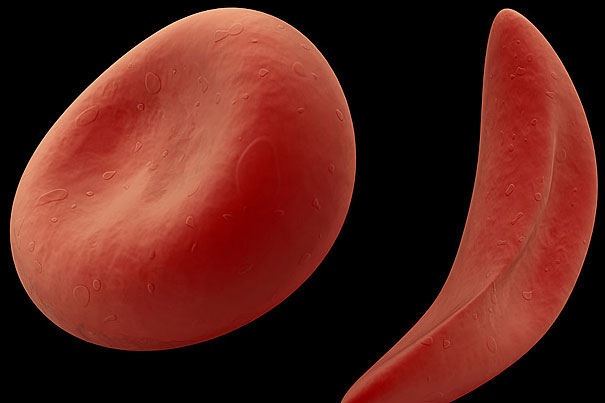
First described more than 100 years ago, sickle cell disease (or sickle cell anemia) is an inherited blood ailment caused by a single mutation in one of the components of hemoglobin, the oxygen-carrying protein in red blood cells. The mutation reduces the protein’s ability to carry oxygen, and forces the cells to curve into a distinctive crescent or sickle shape, causing them to accumulate painfully and break apart in small blood vessels.
BigStock image
Health
Dialing down sickle cell disease
Study in mice says dialing up fetal hemoglobin may bring new therapies
Flipping a single molecular switch can reverse illness in an animal model of sickle cell disease, according to a study by Harvard researchers at Children’s Hospital Boston (CHB) and Dana-Farber Cancer Institute (DFCI). When a protein called BCL11A is turned off, the body is able to manufacture red blood cells with an alternate form of hemoglobin unaffected by the mutation that causes the debilitating illness.
The findings were reported online today in the journal Science by a research team led by Stuart H. Orkin, Harvard Medical School’s David G. Nathan Professor of Pediatrics at the Dana-Farber/Children’s Hospital Cancer Center (DF/CHCC). The results provide strong evidence that BCL11A could be a powerful treatment target for a significant global health problem that affects between 75,000 and 100,000 people in the United States.
“This study provides the first proof of principle that BCL11A might serve as a target for treating sickle cell disease and related blood disorders such as the thalassemias,” said Orkin, associate chief of the division of hematology/oncology at CHB and chair of pediatric oncology at Dana-Farber.
First described more than 100 years ago, sickle cell disease (or sickle cell anemia) is an inherited blood ailment caused by a single mutation in one of the components of hemoglobin, the oxygen-carrying protein in red blood cells. The mutation reduces the protein’s ability to carry oxygen, and forces the cells to curve into a distinctive crescent or sickle shape, causing them to accumulate painfully and break apart in small blood vessels. The disease is found overwhelmingly in patients of African descent.
The disease can only be cured with a bone marrow — blood stem cell — transplant, though difficulties in finding well-matched donors and the complications associated with transplantation rules this option out as an option for most patients. A drug called hydroxurea can provide some relief, but its mechanism of action is unclear and its effectiveness is unpredictable from patient to patient.
Our bodies can manufacture two forms of hemoglobin: the adult form susceptible to the sickle cell mutation, and a fetal form that is largely produced during fetal development and for a short time after birth. Affected children first experience sickle cell symptoms around the time that their bodies switch from producing red blood cells with fetal hemoglobin to those with adult hemoglobin, at about 3 to 6 months old.
BCL11A first drew serious attention in genome-wide association studies of sickle cell disease patients. Shortly after the genomic data came to light, Orkin, who is also a Howard Hughes Medical Institute investigator, and his colleagues showed that by genetically knocking out BCL11A they could activate fetal hemoglobin and silence mutated adult hemoglobin in cultured normal human red cell progenitors.
“The red blood cell lineage performs a balancing act when it comes to manufacturing hemoglobin,” Orkin explained. “Increasing the production of one form reduces production of the other.”
In the current study, Orkin’s team set out to assess the impact of removing BCL11A from the red blood cell lineage in adults. Using two different mouse models of human sickle cell disease as a means of cross-validation, they engineered a system whereby they could selectively turn the protein off just in the immature progenitor cells that give rise to red blood cells.
The results in these mice were striking. Relieved of BCL11A’s influence, the mice in both models started producing red blood cells that showed no evidence of sickling, with no adverse impact on the level of cell production. Eighty-five percent of the red blood cells in the mice carried fetal hemoglobin, and on average 30 percent of the hemoglobin contained within these cells was of the fetal type. The mice also showed great improvement in a number of physiologic and clinical features.
“We knew from previous clinical studies that the body needs to produce cells containing only 15 to 20 percent fetal hemoglobin to reverse disease,” Orkin said, “With these results, we know now we have a target that, if we can develop ways to inactivate or silence it clinically, could be very beneficial to people with sickle cell.”
He and his colleague are already pursuing several lines of investigation aimed at broadening their understanding of BCL11A and translating their mouse findings into potential treatments.
“We want to better understand the network of genes and proteins that interact with BCL11A, to see if any of them might also stand out as targets,” he said. “We also are screening the protein against libraries of chemicals in the hope of identifying compounds that interfere with it, and think it may also hold promise as a target for genetic therapies.”
The study was supported by the National Heart, Lung and Blood Institute, the National Cancer Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, the Howard Hughes Medical Institute, and the Marie Betzner Morrow Endowment.





